Amin Tavakoli
About Me
I am a postdoctoral scientist at Caltech, working with Anima Anandkumar and Frances Arnold in the AI + Science group. My research focuses on developing new machine learning methodologies for scientific discovery, with applications in protein design, chemistry, and quantum physics. I earned my PhD in Computer Science from the University of California, Irvine, where I worked with Pierre Baldi in the AI in Science Institute. During my PhD, I conducted research on developing AI methodologies for molecular design, reaction prediction, and computational modeling of chemical processes, as well as advancing the theoretical foundations of deep learning.
Education
- Ph.D. Computer Science, University of California, Irvine, 2023
- B.Sc. Physics & B.Sc. Engineering, Sharif University of Technology, 2017
Research Interests
Ordered by the extent of my current involvement.
- Computational Protein Design - Protein Language Models
- Generative Modeling of Molecular Systems: From Small Molecules to Proteins
- Geometry Aware Deep Learning
- Foundational Models for Quantum Chemistry
- Brain-inspired Learning Algorithms
Publications
You can find my publications here: Google Scholar
Recent Projects
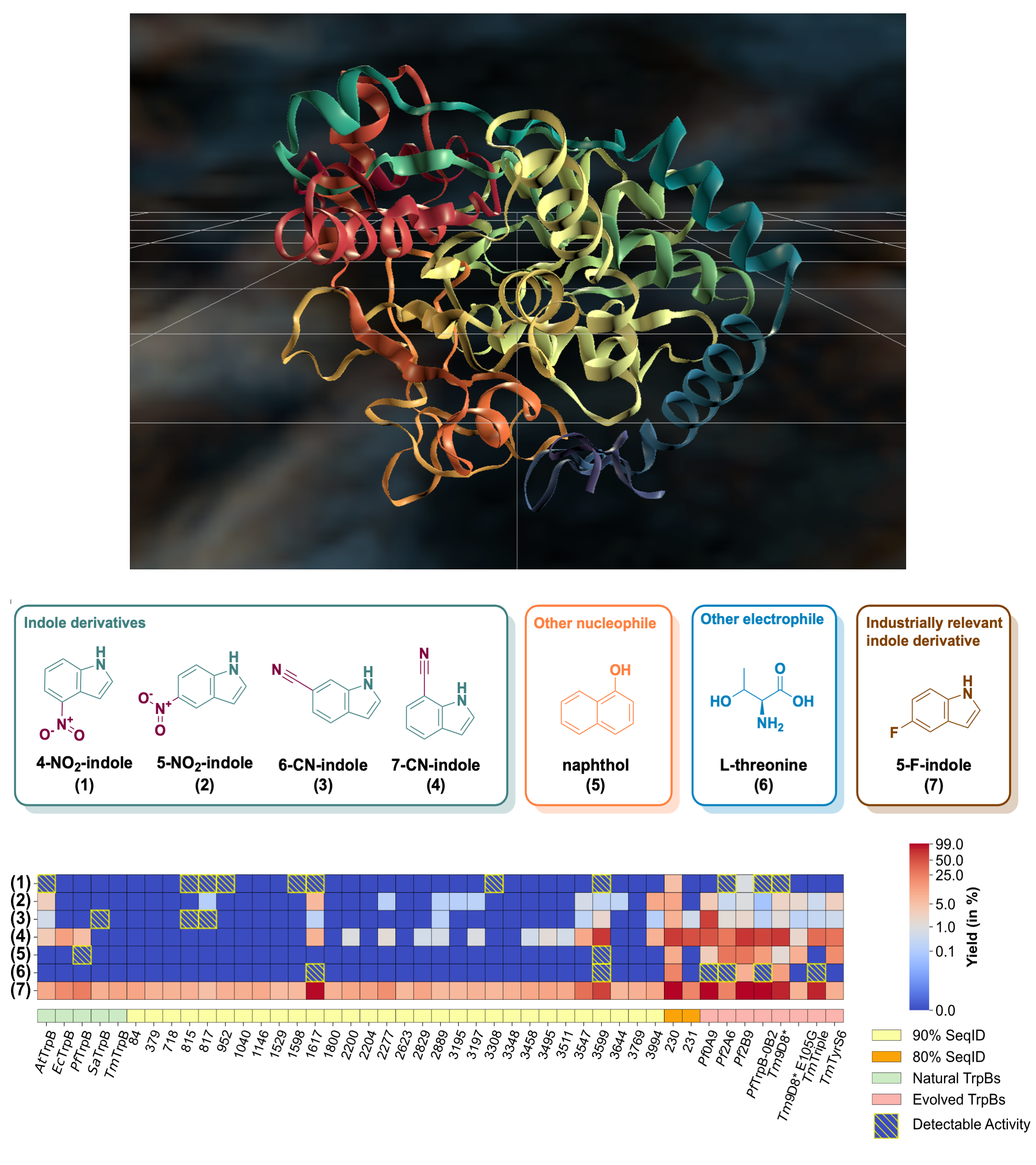
We use the GenSLM protein language model (PLM) to perform sequence-conditioned generation of TrpB enzyme variants, addressing a major challenge in biocatalyst design: finding functional starting points for optimization. By combining generative modeling with computational filtering, the approach produces sequences that are stable, expressible, and catalytically active, some operating independently of their natural partner. Several AI-generated variants outperformed both natural and lab-optimized enzymes, showing that the model captures latent functional patterns and can propose non-trivial, high-performing sequences beyond evolutionary examples. This demonstrates that PLMs can act as powerful generative priors for functional protein engineering, enabling AI-driven exploration of functional sequence space with minimal experimental iteration.
PDF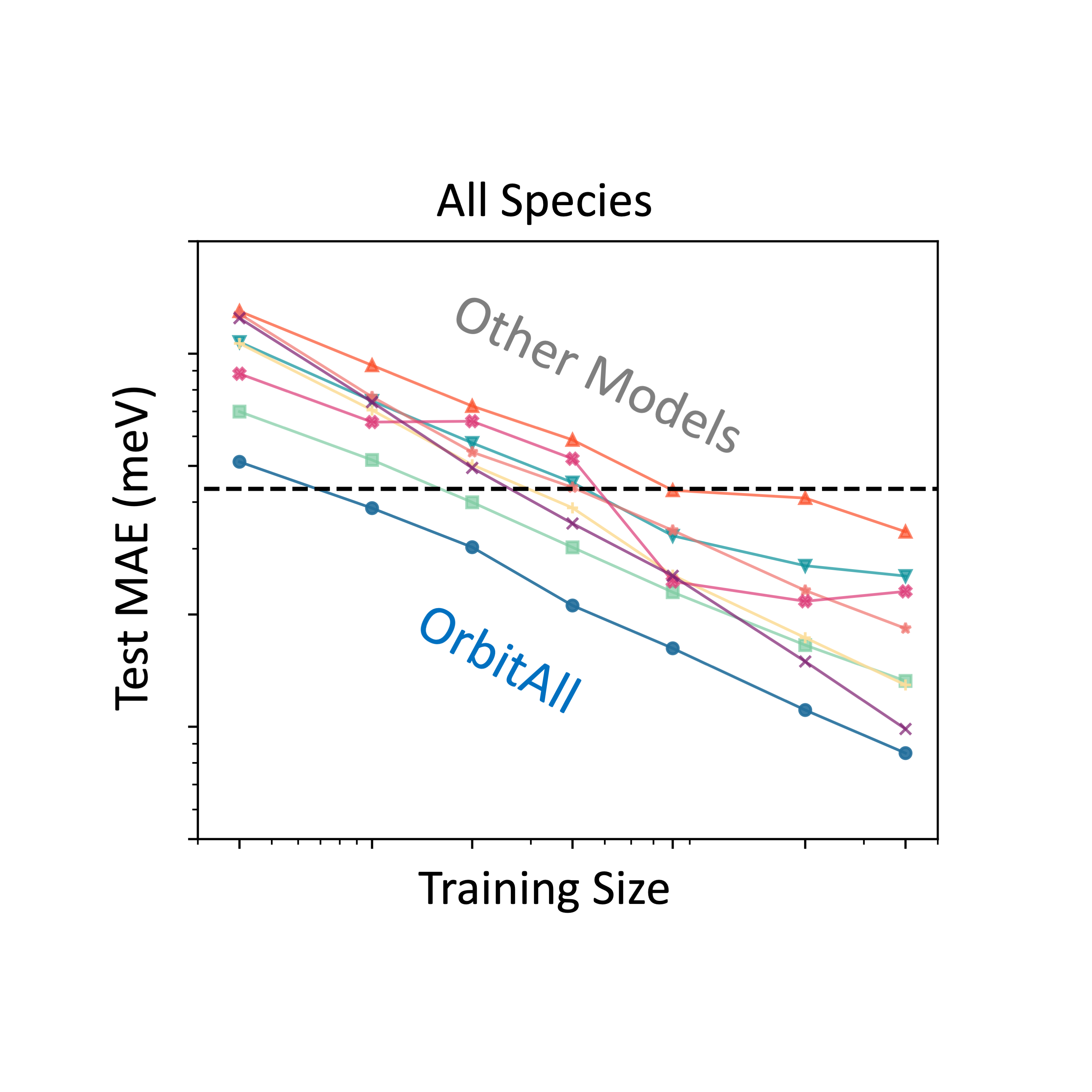
While deep learning has advanced quantum chemistry, most models remain limited to neutral, closed-shell molecules. In contrast, real-world systems involve varying charges, spins, and environments. We present our model—a geometry- and physics-informed deep learning framework that incorporates spin-polarized orbital features and SE(3)-equivariant graph neural networks to represent arbitrary molecular systems. Our model accurately predicts properties of charged, open-shell, and solvated molecules, and generalizes to much larger systems than seen during training. It reaches chemical accuracy with 10× less data than competing models and offers a 1,000–10,000× speedup over DFT, thanks to its physics-grounded design.
PDF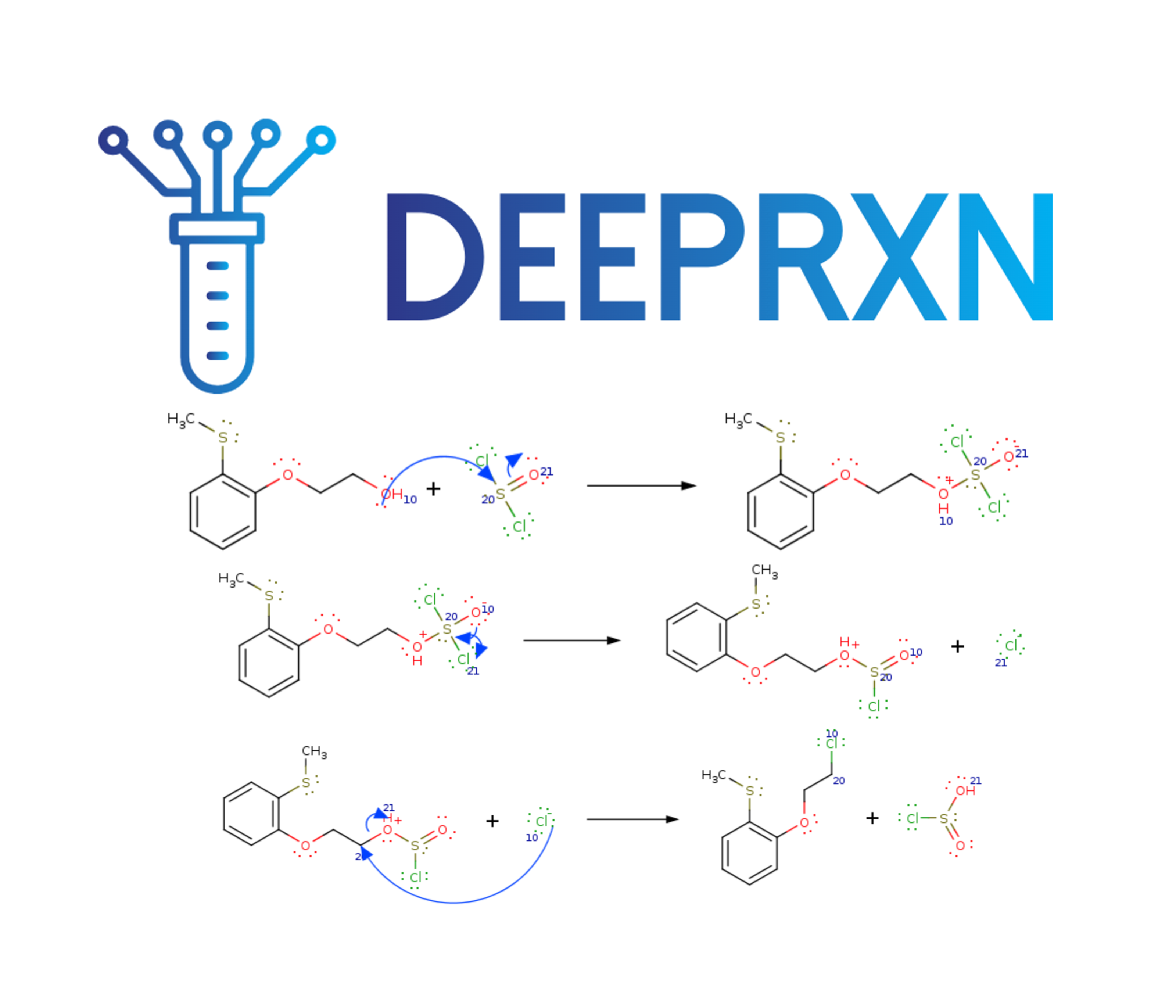
DeepRXN is a specialized platform designed to advance the integration of deep learning into chemoinformatics, hosting predictive chemoinformatics software and public chemical reaction databases. Its unique focus on representing chemical reactions through elementary step reactions offers numerous advantages, with applications spanning reaction prediction, synthetic planning, atmospheric chemistry, drug design, and beyond. This innovative perspective holds the potential to reshape and elevate various aspects of chemical research and applications within a singular framework.
PDF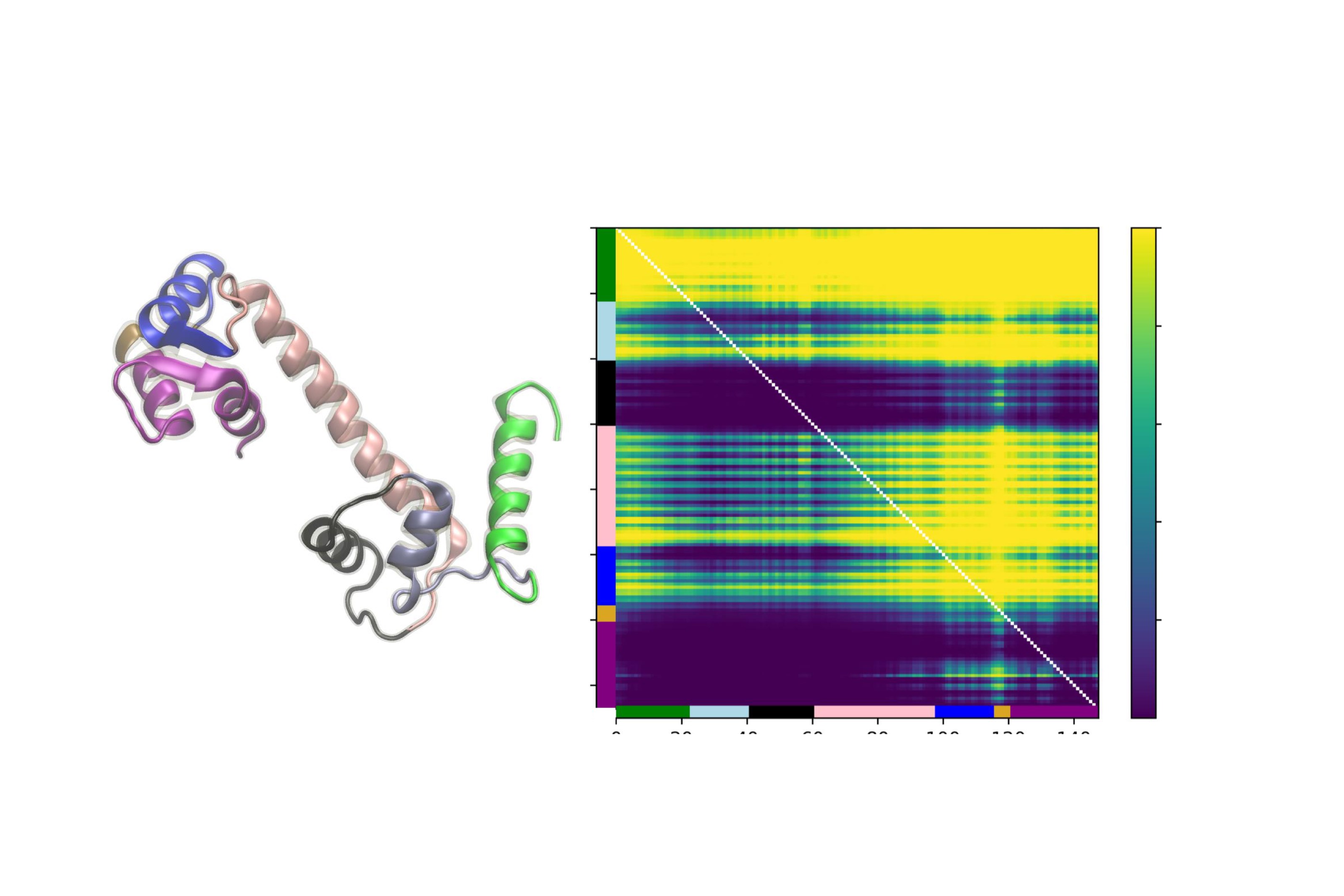
Machine learning models for protein engineering typically use sequence-based, structure-based, or combined representations rooted in the idea that sequence and structure inform function. While effective in capturing evolutionary patterns, these approaches often overlook protein dynamics. We introduce a dynamic-aware representation derived from unsupervised analysis of molecular dynamics simulations. By encoding temporal and spatial behaviors, our method captures key interactions within the protein chain, offering valuable insights for protein design.
PDF
Teaching & Mentoring
- Advising three undergraduate students on computational biology and quantum chemistry projects.
- Teaching non-eucleadian deep learning at UCI data science workshop.
- Teaching deep learning and neural networks (CS274) as the invited lecturer.
Mentees:
- Raswanth Murugan - Caltech 2025
- Beom Seok Kang - Caltech 2024
- Ian Domingo - UCI 2023
- Ryan Miller - UCI 2023
- Mirana Clair Angel - UCI 2023
Good Reads (from everywhere!)
Contact
Last update: Oct 2025